
Silicon carbide (SiC) is a very promising semiconductor material for high-temperature, high-frequency and high-power optoelectronic devices. Among SiC polytypes, its amorphous alloy form (a-SiC) combines several features of great technological interest: diffusion barrier performance, tunable bandgap, p- and n-doping, conductivity, source of hydrogen and dopants, surface passivation, thermal stability and stress tuning. Amorphous SiC is used as protective coating against corrosion, etching and abrasion, for bio-molecular and medical applications, as well as in the production of solar cells, photoreceptors, X-ray and color sensors, and visible light LED.
The Challenge
The immense interest in SiC materials is due to the possibility of tailoring the structural, mechanical, and optical properties by changing the degree of amorphization. As such, the development of computational strategies that accurately model the atomistic structure and provide insights into the structural, mechanical, and electronic properties of a-SiC is of great importance for the design of materials for specific applications. Complete description of an amorphous structure requires the knowledge of the chemical and topological order, assessed by structural properties, such as chemical (dis)order and pair distribution function, and directly correlated with the mechanical performance. SiC has a very widely tunable stress parameter range. Molecular dynamics (MD) simulations are well-suited for obtaining reliable atomic structure of a-SiC by quenching melt approach, further required for predictions of mechanical properties, such as the Young’s moduli.
The Work
Molecular dynamics simulations using the LAMMPS plugin in MAPS, coupled with a Tersoff reactive potential containing parameters for Si and C, were used to generate the a-SiC material from 3C-SiC crystal structure. Several analysis tools within MAPS platform were used to characterize the a-SiC structural properties. The Young’s moduli were computed on the initial linear part of the stress/strain curve using MAPS mechanical properties analysis.

Figure 1: Initial (a) and final (b) conformation of a-SiC after subjecting it to a mechanical compression along the z axis.
The Results
Crystalline SiC in a cubic box with a lattice parameter of 21.84 Å and 512 atoms in a zinc-blended structure was constructed. The structure was first heated up to 6000 K for 200 ps in order to randomize the initial conformation of the silicon carbide. Then, the system was gradually quenched down to 300 K during a 6 ns NVT run, followed by an NPT equilibration at 300 K for 500 ps. Similarly, Si and C diamond crystalline systems (50 x 50 x 50 cubic cell), used for comparison, were constructed and rapidly equilibrated at 300 K over 200 ps.
Structural Properties. The chemical disorder parameter () was computed for the final conformation of the equilibration run of a-SiC. The c (resp Si) is defined for C (resp Si) atoms as NC-C/NC-Si (resp NSi-Si/NC-Si), where NC-C is the number of C-C bonds (resp NSi-Si the number of Si-Si bonds) and NC-Si the number of C-Si bonds. This parameter is a marker of the system disorder, and it ranges from 0, for a completely ordered to 1, for a completely disordered system. The two disorder parameters were found to be similar around C = Si = 0.48, which is in very good agreement with results of Xue et al. [1].
The a-SiC material was further characterized by analysis of the pair distribution functions, computed for C-C, Si-Si, and C-Si atom pairs (see Figure 2). The pair distribution function shows a maximum around 1.47 Å for C-C pair, 1.85 Å for C-Si pair, and 2.35 Å for Si-Si pair, in excellent agreement with previous theoretical data [1].
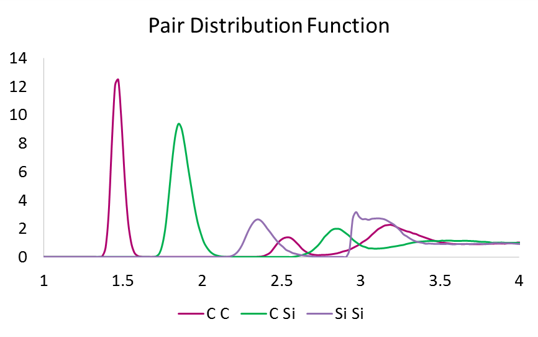
Figure 2: Pair distribution function for C-C, C-Si and Si-Si atom pairs.
A 2x2x2 supercell of the final conformation of the 300 K equilibration (49 x 49 x 49 cubic cell) was generated for the mechanical property simulations. A compression of the c lattice parameter up to 75 % of its initial value (from 49.0 Å to 36.7 Å) was obtained in an NVT run for 5 ns, in which the a and b lattice parameters were allowed to freely equilibrate. The initial and final structures of this simulation are represented in Figure 1-a and b. A similar simulation protocol was used for the Si and C diamond crystalline systems. Additionally, a higher accuracy simulation was performed for the a-SiC material allowing up to 95 % compression of the c lattice over a 10 ns simulation NPT run.
Mechanical Properties. The Young’s moduli of a-SiC, Si, and C diamond are shown in Table 1. The molecular dynamic simulations using Tersoff potentials allowed to reproduce accurately experimental values of the Young’s moduli of silicon and carbon diamond crystals. Moreover, the Young’s modulus of a-SiC materials was found to be lower than that of 3C-SiC system and in good agreement with previous results [2,3].
| Materials | Young’s modulus (GPa) | |
| Computed | Experimental | |
| a-SiC | 325 | 260 – 372 |
| Si | 125 | 130 – 185 |
| C diamond | 1072 | 1050 – 1210 |
The simulations presented here are important for the prediction of structural and mechanical properties of solid materials as they can validate the experiments and guide the search of novel materials with desired properties.
[1] Xue K., Niu L.-S., Shi H.-J., J. App. Phys., 2008, 104, 053518.
[2] Xue K., Niu L.-S., J. App. Phys., 2009, 106 (6), 083505.
[3] Ivashchenko V.I., Turchi P.E.A., Shevchenko V.I., Phys. Rev. B, 2007, 75 (8) 085209.