In numerous technical applications gas adsorption plays a crucial role. One prominent example is the removal of gases from steams. Other applications concern alternative fuel storage and gas separation. Increasingly stricter norms regarding greenhouse gas emission and enhanced oil recovery are driving forces for a prospering carbon capture and storage market. The latter is predicted to grow by over 9% within the next few years. Increasing the adsorption capacity of sorbent materials is therefore a major task. Carbon nanotubes (CNTs) and activated carbon are discussed as promising solid sorbents due to their availability, tunable pore structure and surface chemistry, and the low energy requirements for regeneration.
The Challenge
A promising solid absorbing material has to exhibit a variety of unique physical and chemical properties. Various parameters, such as interactions between different phases, pore geometry or thermodynamic conditions, control the selective adsorption. In this study, we used MAPS Towhee engine to investigate the adsorption of the pure gases CO, CO2, N2 and CH4 in CNTs and the absorption capacity of activated graphite as a function of the density of active sites to enlighten how the adsorption capacity of these materials can be optimized through surface modifications.
The Work
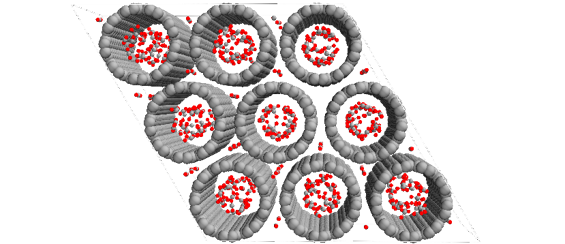
Suitable model systems were generated using MAPS building tools. First, the excess chemical potential of each pure gas was calculated at the thermodynamic state points of interest by means of NPT Monte Carlo (MC) simulations using Towhee. In a second step, grand canonical MC (GCMC) simulations were performed to determine absorption of these gases in the carbon nanotube models. Absorption isotherms were calculated at 298 K for a pressure range between 0.01 – 2.0 MPa. Furthermore, the absorption of CO2 and CO on activated graphite was studied. In order to activate the graphite model, carboxyl and hydroxyl groups were introduced at three different surface densities.
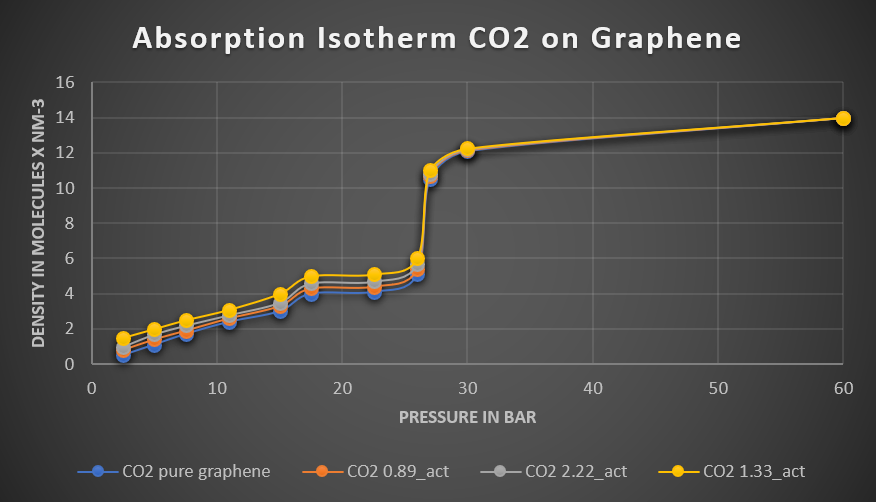
The Results
The simulated amount of the absorbed gases decreases in the order CO2 > CO > N2, CH4. The observed trend is in agreement with experimental findings. Moreover, it was found that the absorption capacity of graphene increases with the density of the activation sites. The analysis of the density profiles obtained from the simulation shows that most of the gas absorbed molecules are distributed near the pore wall indicating a capillary condensation.
The presented approach can be applied in general to study adsorption in porous materials and represents a useful strategy to systematically study the impact of surface modifications.