
Alkane cracking, i.e. the breaking of larger hydrocarbons into smaller, more useful alkanes, is of main importance for producing feedstock for petrochemical industry. Petrochemicals are used across almost all market sectors and the global market value is several hundred billion USD. While thermal cracking involves very high temperatures (500 – 900 °C) and pressures (7000 kPa), catalytic cracking uses eco-friendly, non-toxic zeolites, which are hydrated aluminosilicates possessing a very porous crystalline structure. Fluid catalytic cracking requires intrinsically less energy for alkane dissociation reaction and belongs to major conversion processes in refineries.
The Challenge
Catalytic reactions are involved in the vast majority of chemical production processes at some stage. There is ongoing need to develop and improve catalysts for pure economic, but also environmental reasons. Production processes need to be optimized to lower costs. Apart from that, there exists a rising consciousness with respect to sustainable and environmentally friendly manufacturing. Key for achieving higher yields and optimizing reaction conditions is the knowledge of the catalytic mechanism. In this context, materials simulations can provide critical insights, because it allows to investigate situations that are experimentally inaccessible or difficult to realize, e.g. transition states to mention only one example.
The Work
In the present case study, we performed Density Functional Theory (DFT) calculations using the MAPS/NWChem module for investigating the zeolite catalyzed propane dissociation into ethylene and methane molecules. We focus on the understanding the origin of the catalytic effect and the influence of different parameters such as temperature and pressure on the catalytic rate of the reaction. The reaction profile was calculated, and transition states were identified for different steps. Moreover, charge calculations have been performed and MAPS Thermodynamic analysis tool was used to determine the influence of temperature and pressure on the reaction energies.
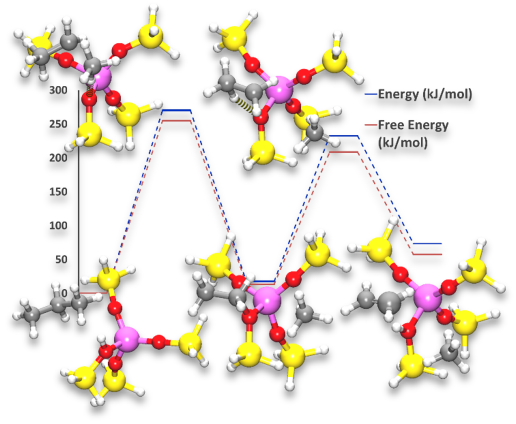
Figure 1: Energy evolution along the reaction path of propane cracking.
The Results
The catalytic cracking process of propane into ethylene and methane involves two steps. The calculated reaction energies reveal that the first step is the rate determining one, because the corresponding energy barrier is ~100 kJ/mol higher compared to the second step. This finding is in accordance with literature date. The analysis of the reaction pathway shows that the zeolite induces the formation of a stable intermediate from which a methane molecule is created and the C2H5 group is bonded to the zeolite. This intermediate appears very stable and therefore induces a lowering of the two barriers. The analysis of the atomic partial charges indicates that the catalytic effect of the zeolite is due to the proton acidity that induces a polarization of the C-C bond which in return stabilizes the formation of the intermediate state.
Molecular modeling is a versatile tool to understand and predict molecular reactivity for complex systems which can be crucial in catalysis, oil & gas and chemical industries.
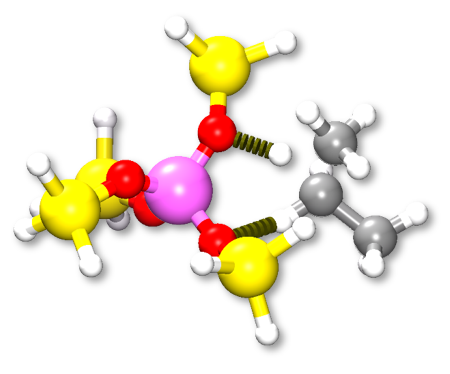
Figure 2: 3D structure model of transition state of first reaction step.