
Microporous materials are of great economic importance and play a crucial role as catalysts, molecular sieves, and for clean energy production and storage. Carbide-derived carbon (CDC) forms a class of porous materials which is regarded as a superior electrode material for energy storage devices like supercapacitors. Tunable pore size and a large surface area make CDCs to promising key materials for electrodes used in energy storage devices.
The Challenge
Amorphous porous systems are used in a variety of applications and market opportunities are still expanding, but this kind of materials are often very difficult to characterize fully through experiment. A thorough understanding is, however, essential for being able to optimize and tune product performance. Nanoporous carbon is regarded as one of the most promising electrode materials for supercapacitors. In this project, we have studied how the precursor material and impurities affect the porosity, sorption behavior and the carbon 3D structure, which is fundamental for the chemo-physical properties. The goal is to create realistic digital twins of the final material for studying charging processes and ultimately increasing the capacitance of supercapacitors.
The Work
The structure of amorphous nanoporous carbon systems and their adsorption behavior were modeled using MAPS/LAMMPS and MAPS/Towhee modules. MAPS analysis functionalities were used to characterize the structures. Different carbide and carbonitride precursors were subjected to a set of molecular dynamics simulations (MD) for creating realistic models of the activated electrode material. Pore size and sp3/sp2 ratio were analyzed. CO2 adsorption isotherms were calculated for pure carbon and partially oxidized carbon models.
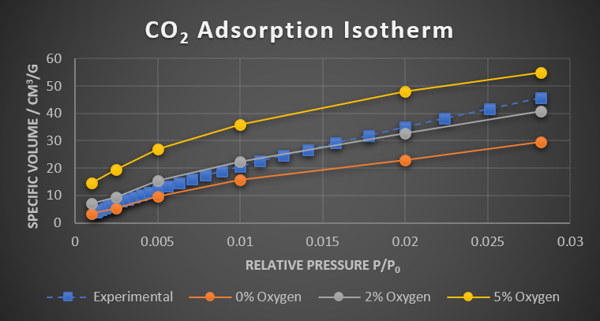
Figure 1.Calculated and experimental CO2 adsorption isotherms.
The Results
We have established a pseudo-experimental simulation protocol capable to generate amorphous nanoporous structures. This is fundamental for studying properties and processes at the atomistic level. Structures were validated by comparing calculated properties with experimentally determined data. The simulations showed that experimental structure contain 2% oxygen impurities. The simulation protocol is generally applicable for modeling amorphous porous materials and also been applied successfully in a slightly modified form for studying an amorphous heterogeneous catalyst system.
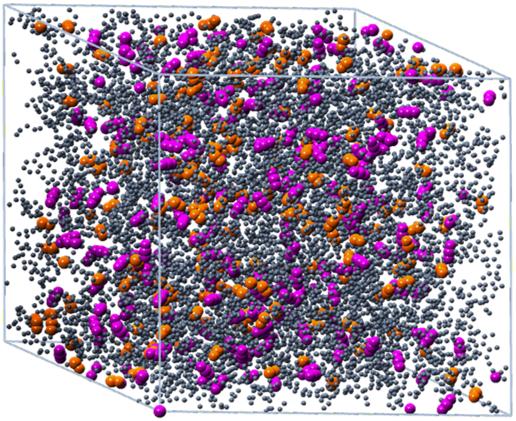
Figure 2: Model of partially oxidized nanoporous carbon Color code: carbon skeleton – gray, C=O groups – orange, CO2 – magenta