
Perlfluorocarbon-based nanoemulsion particles (PFC-NEP) have become promising platforms for the delivery of therapeutic and diagnostic agents to specific target cells in a non-invasive manner. This nanoparticle platform is capable of multifunctional ligand/drug interactions including peptides, small molecules, biological and antibody cargos, and thus may represent an economical platform for polyvalent targeting and multiplexing therapeutics. The proposed mechanism for this “contact-facilitated” delivery starts with the formation of a hemifusion complex between the monolayer of the PFC-NEP and the outer monolayer of the target cell plasma membrane. The cargo molecules then diffuse to the plasma membrane through the hemifusion complex to become internalized by lipid raft mediated endocytosis. The molecular details of this mechanism are however not fully understood, hampered by the complexity of the required experiments.
The Challenge
Computational experiments by means of molecular dynamics simulations at both atomistic and mesoscopic levels can be extremely valuable at assessing such a complex mechanism. Atomistic simulations can provide accurate determination of structural details, but become computationally costly at examining the larger-scale membrane behavior. On the other hand, mesoscopic models offer the advantage of simulating large systems at long time-scales by reducing the number of degrees of freedom as compared to the all-atoms models.
The Work
In the following case study, we simulate at a mesoscopic level i) the spontaneous formation of PFOB-NEP particles, ii) the self-assembly of lipids into bilayers and vesicles, and iii) membrane fusion between PFOB-NEPs and phospholipid vesicles. The MD simulations were performed using the GROMACS plugin within the MAPS platform and employed the Martini force field [1], which has been used for large variety of biomolecular processes.
PFOB is modelled by three interaction sites: BRC (BrCF2CF2CF2–), CBM (–CF2–CF2–CF2–), and CBE (–CF2–CF2–CF3) such that the mesoscopic model is a linear 3-particle system, BRC–CBM–CBE. Bond length is determined based on the Lennard-Jones (LJ) size parameters and bending angle is based on the existing Martini representation of the mesoscopic lipid aliphatic chain. The three sites are modelled by a new bead type, which was developed in Lee et al. [2] to preserve the stronger hydrophobic character of perfluorocarbons as compared with hydrocarbons. In particular, the initial guess for the LJ parameters (σ=0.47 nm, ε=3.5 kJ/mol) is taken from the apolar bead type C1 of the Martini force field, and the final optimized parameters reproducing the density and heat of vaporization of liquid PFOB are σ=0.5 nm, ε=3.5 kJ/mol. The interaction of PFOB with other molecules (lipids and water) is described using the constant size parameter of σ=0.5 nm, and different well depth parameter, ε, based on the parameterized level of attractiveness between the two sites.
The Results
Spontaneous formation of PFOB-NEP particles
The PFOB-NEP droplet was generated by randomly mixing of 169 POPC and 170 POPE phospholipids with 1000 PFOB molecules and 320,000 water molecules (80,000 P4 beads) in a box with dimensions 25.3 nm x 25.3 nm x 25.3 nm. Energy minimization was followed by an isobaric-isothermal ensemble (NPT) run at a temperature of 303 K. Figure 1 illustrates snapshots of the NPT simulation starting from a random mixture and gradually forming PFOB-NEP droplets.
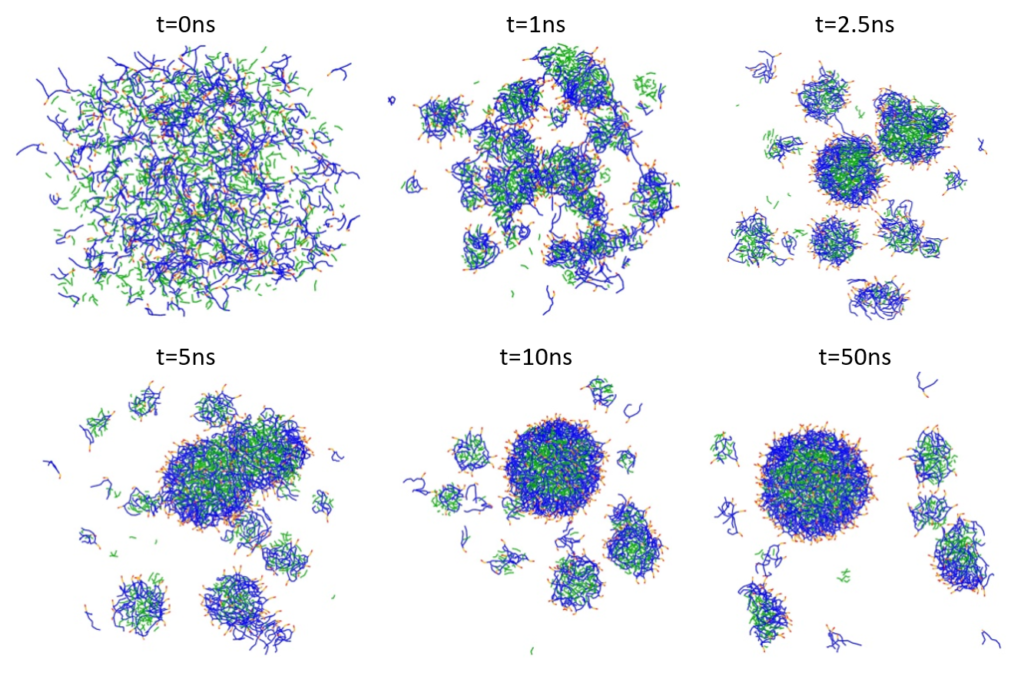
Figure 1: Spontaneous emulsion formation. PFOB is shown in green, lipid tails in blue, and lipid polar head groups in red and orange. Water particles are omitted for clarity.
Liposome formation
The liposome formation was performed according to the protocol of Marrink and Mark [3]. Figure 2 shows the spontaneous formation of a phospholipid bilayer starting with a random mixture of 128 POPE, 127 POPC and 3,000 P4 beads (or 12,000 water molecules) within an NPT run at T = 303 K.

Figure 2: Spontaneous phospholipid bilayer formation. Lipid tails are shown in blue, and lipid polar head groups in red and orange. Water (P4) particles are displayed in cyan.
The bilayer was then placed in a simulation box and water molecules were added to solvate the bilayer in all directions. Energy minimization using the steepest descent method was followed by an NPT run at T = 303 K. The liposome formation starting from a POPC/POPE bilayer, which gradually bends, encapsulates water molecules and finally closes to form the vesicle structure is illustrated in Figure 3.
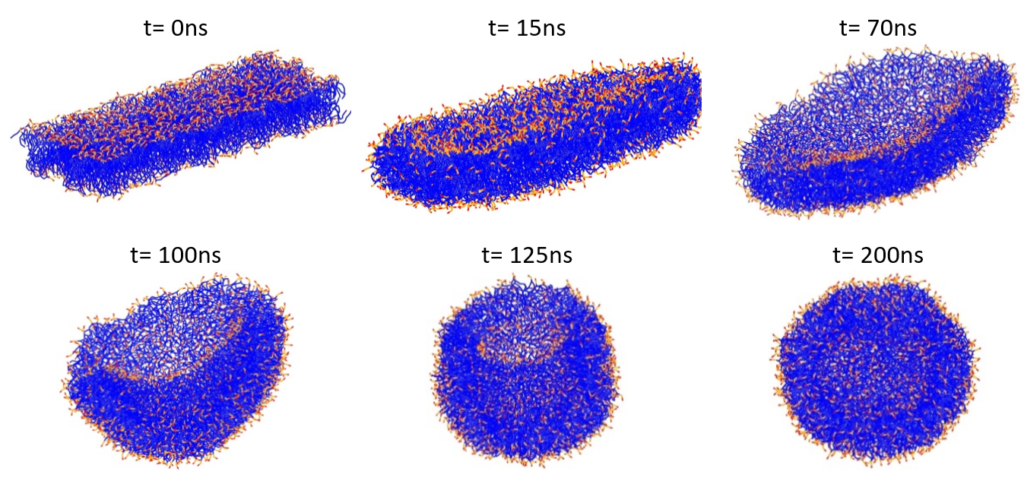
Figure 3: Formation of a phospholipid liposome starting from a POPC/POPE bilayer. Lipid tails are shown in blue, and lipid polar head groups in red and orange. Water particles are omitted for clarity.
Liposome / PFOB-NEP fusion
The fusion simulation between the liposome and the PFOB-NEP particles was performed by placing the two spherical structures at a distance of less than 1 nm between their closest lipids and surround them with water molecules. The thin water layer (approximately 3nm) between the two particles was removed and a short MD simulation was performed at constant pressure. The vacuum slab induced (a) the reorientation of lipid molecules, leading to exposure of lipid tails to the vacuum region driven by the hydrophobic effect and (b) a transient low pressure between the spherical structures, creating an attraction between them. Lipid reorientation and close contact between the structures induced the formation of a stalk, which quickly transformed into a hemifused state. The initial configuration and the stalk formation are shown in Figure 4(a) and (b), respectively.

Figure 4: (a) Initial configuration of PFOB-NEP and phospholipid vesicle. (b) Re-orientation of lipid molecules leading to stalk formation. The lipid tail (in blue) of one of the vesicle molecules gets exposed (red circle). Lipid polar head groups are in red and orange, and PFOB molecules are in green.
The hemifusion complex signals the delivery of the cargo PFOB molecules, enabling their diffusion to the plasma membrane of the target liposome and their gradual internalization by lipid raft mediated endocytosis. This process is illustrated in Figure 5.
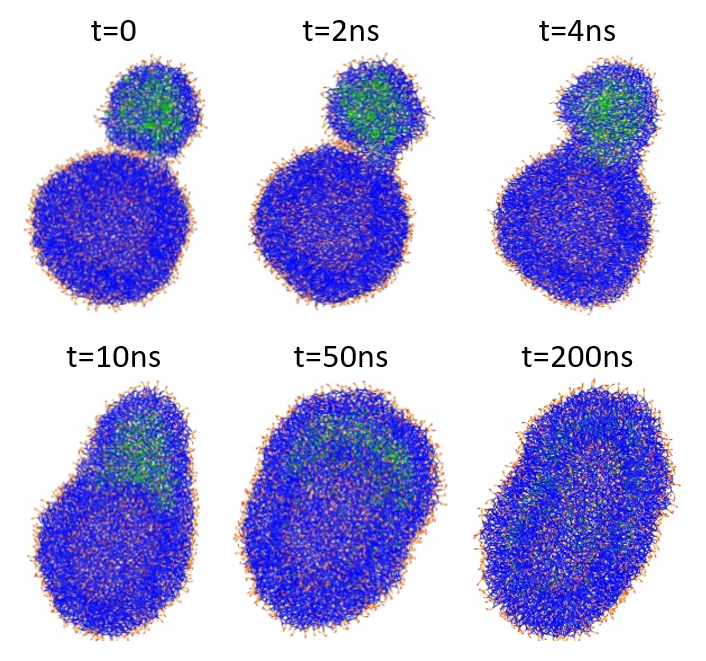
Figure 5: Structural changes during the fusion of a PFOB-NEP particle and a phospholipid vesicle. PFOB is marked in green, lipid tails are in blue, and lipid polar head groups are in red and orange.
The presented approach can be used for systematic analysis testing the fusion dependency on particle size and lipid dependency, allowing better characterization of the involved mechanisms, as well as the design of particles for optimal delivery.
- Marrink, S.J., Risselada, H.J., Yefimov, S., Tieleman, D.P., & De Vries, A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B, 2007, 111, 7812-7824.
- Lee, S.J., Schlesinger, P.H., Wickline, S.A., Lanza, G.M., & Baker, N.A. Simulation of fusion-mediated nanoemulsion interactions with model lipid bilayers. Soft Matter, 2012, 8, 7024-7035.
- Marrink, S.J., & Mark, A.E. Molecular dynamics simulation of the formation, structure, and dynamics of small phospholipid vesicles. J. Am. Chem. Soc., 2003, 125, 15233-15242.